金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
外显子组测序鉴定了来自听力障碍、甲营养不良和癫痫的两个无关家庭的三个候选基因的变异

【关键词】DDOD(耳聋-甲营养不良)、听力障碍、全外显子组测序(WES)、ATP6V1B2、TJP2、KIF11

为了调查先天性听力障碍的遗传原因,对166个先天性耳聋家庭中的542名个体进行全外显子组测序,在两个无关家庭的五名受影响个体中鉴定出三种变异。家族1中,在患有DDOD的母亲和儿子中发现ATP6V1B2基因的无义突变(c.1516C>T,p.R506*);同时,在患者中检测到TJP2基因中一个新的杂合变异(c.1590T>G,p.D530E)。家族2中,在患有DDOD的父亲和女儿中检测到ATP6V1B2基因的相同突变;此外,女儿的母亲患有感音神经性耳聋和癫痫,在她身上发现了KIF11基因中的一个新的杂合变异(c.733A>G,p.M245V)。本研究主要结果:(1)首次证明ATP6V1B2基因引起的DDOD综合征是一种常染色体显性遗传病;(2)KIF11基因中的变异(c.733A>G,p.M245V)位于驱动蛋白马达结构域中,推测该区域可能参与包括耳蜗细胞在内的中枢神经系统中的蛋白质相互作用。临床价值:扩大了听力障碍候选基因的变异谱,并对ATP6V1B2、TJP2和KIF11基因对听力功能的作用提供了新的见解。详细的基因型-表型分析可以进一步表征听力障碍相关变异和潜在发病机制。

听力障碍是人类最常见的遗传缺陷之一1,分为综合征性听力障碍(SHL)和非综合征性听力障碍(NSHL)。共鉴定出124个导致NSHL的基因。大约30%的听力障碍患者被认为是综合征患者2,目前描述了400多种不同的听力障碍综合征3。DDOD综合征(MIM:220500)的病因首次从3个无关病例中发现ATP6V1B2基因中的新发杂合变异(c.1516C>T;p.R506*)4。在具有更广泛临床表现的患者中也检测到该变异,包括耳聋、甲营养不良、智力残疾(智力迟钝)或癫痫(称为DOORS综合征)5。这是在ATP6V1B2基因中发现的唯一LoF变异。然而,自1961年首次发现DDOD综合征以来,这种LoF变异(c.1516C>T,p.R506*)是否与常染色体显性遗传有关尚不确定6。


本研究招募了两个无关家庭中的五名受影响个体。对有听力障碍和其他症状的患者进行常规临床试验。收集个人和家族史,包括听力障碍、耳鸣、前庭症状、氨基糖苷类药物的使用和其他异常情况。对受试者进行全外显子组测序和生物信息学分析,排除在gnomAD或内部中国外显子组数据库中检测到的MAF>0.001的变异,使用特异性引物进行Sanger测序,并根据ACMG指南对变异进行致病性评级。

临床表型—家庭1
先证者是6岁的男孩(图1A、B),患有双侧深度感音神经性耳聋,于20个月龄时接受单侧人工耳蜗植入术后,听力和言语能力恢复正常。第1和第5指指甲缺失,食指指甲再生性障碍,其余手指的指甲表面粗糙不平。拇指呈指状,第5指异常短。X光显示双侧食指远端指骨缺失(图1C、D)。右脚第1、2趾无趾甲,第3、5趾严重发育不良,左脚第1、3趾无趾甲,4、5趾趾甲残缺和粗糙。足部检查和X线片显示第5趾远端双侧发育不良(图1E、F)。先证者的母亲(图1G)也表现出类似的表型,如双侧深度先天性感音神经性耳聋和甲营养不良(图1H、I)她的脚趾甲发育受到严重影响,(图1J、K)。
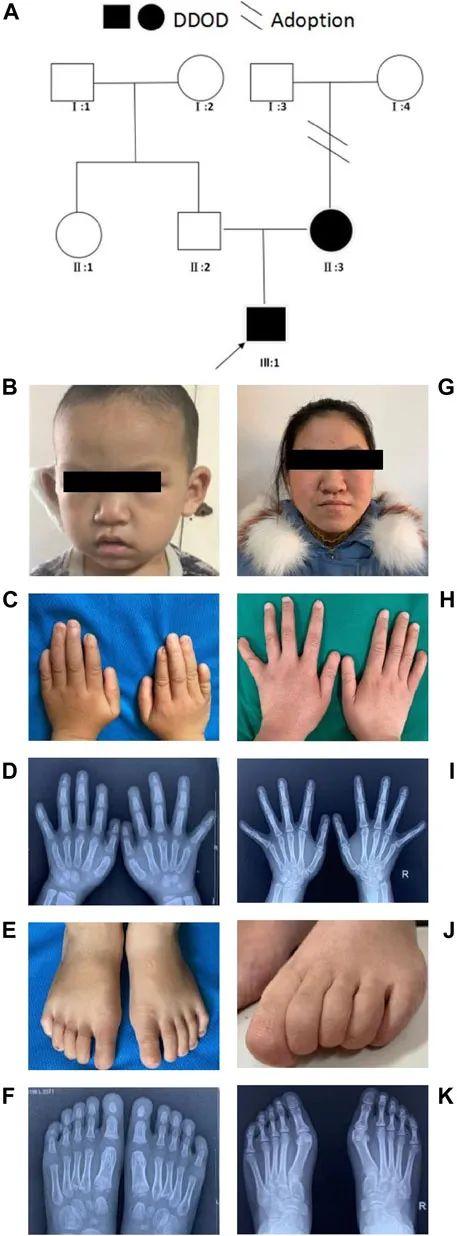
图1 家庭1的临床表型
临床表型—家庭2
先证者是7岁的女孩(图2A、B),患有双侧重度感音神经性听力障碍。3岁时,她接受了单侧人工耳蜗植入。先证者不善于主动表达,记忆力差。体检发现她的手指和脚趾出现双侧异常(图2C-F)。第1、3指指甲缺失,其余手指明显发育不良。拇指呈指状,左手五指异常短。脚趾甲完全缺失。她的脚是平的(扁平足)。牙科检查显示牙齿发育不全。先证者的父亲(图2G)表现出与女儿相似的表型,包括严重的先天性感音神经性听力障碍和甲营养不良(图2H、I)。此外,他的足部X光片显示右脚第2脚趾远端发育不良(图2J、K)。先证者的母亲被诊断患有严重的先天性双侧感音神经性耳聋、癫痫和轻度智力残疾。
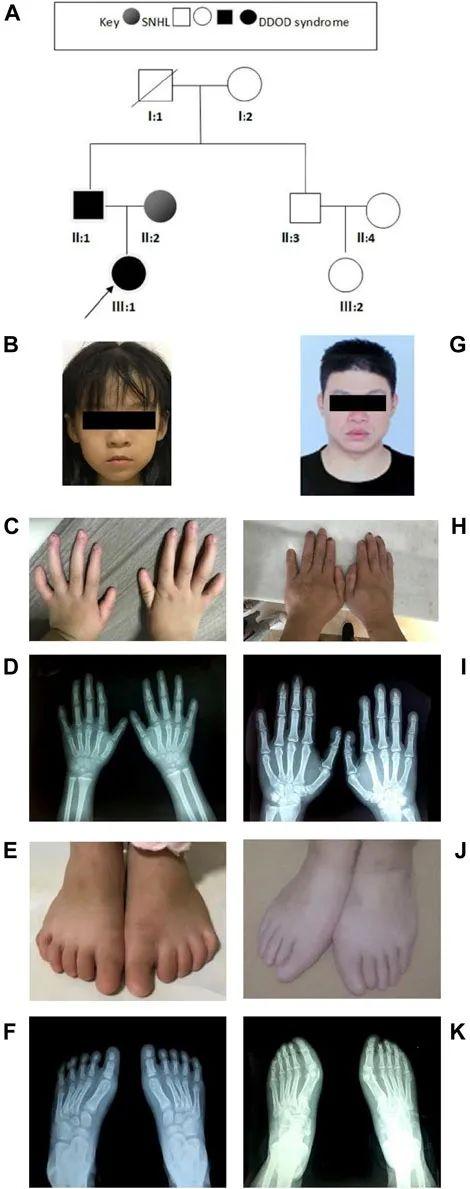
图2 家庭2的临床表型
ATP6V1B2基因中致病性变异的鉴定
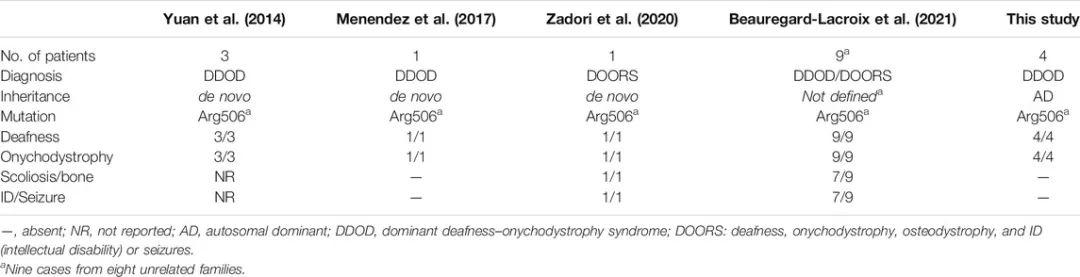
在两个家庭的四名有听力障碍和甲营养不良的患者中发现ATP6V1B2基因的一种已知的致病性变异(c.1516C>T;p.R506*),该变异先前被报道为多例耳聋和甲营养不良以及多例DOORS综合征的新发变异。在这两个家庭中,该LoF变异可以遗传给下一代,与常染色体显性遗传病的形式一致。根据ACMG指南,该变异为PVS14、PP3、PP5和PP1。因此,被归类为致病性变异。目前,在HGMD中已发现ATP6V1B2基因的7种不同变异,只有位于c末端区域的LoF变异(c.1516C>T,p.R506*)与听力障碍和甲营养不良相关4。在本报告中,ATP6V1B2基因中的LoF变异与两个无关家族中的四名受影响个体共分离,表明c.1516C>T变异是常染色体显性DDOD综合征的遗传原因。几乎先前报告的c.1516C>T(表1)病例都是由该等位基因新发突变引起的。
表1 ATP6V1B2基因中c.1516C>T变异导致的DDOD/DOORS患者比较
在家庭1的患者中检测到TJP2基因的变异
家庭1中受影响的母亲和儿子在TJP2基因中携带了一个变异(c.1590T>G,p.D530E,图3A)。TJP2基因是肝内胆汁淤积症和感音神经性耳聋的已知候选基因7。在小鼠胚胎中,TJP2在肝脏、胆管、连接毛细胞和支持细胞的耳蜗膜中高度表达8,表明其在肝脏和耳蜗发育中的作用。在HGMD中,TJP2的59种不同变异与多种疾病有关,如肝内胆汁淤积症、高胆固醇血症和听力障碍。编码蛋白中分散了10种与听力障碍相关的变异(图3B)。在我们的案例中,c.1590T>G(p.D530E)变异位于PDZ3结构域中。
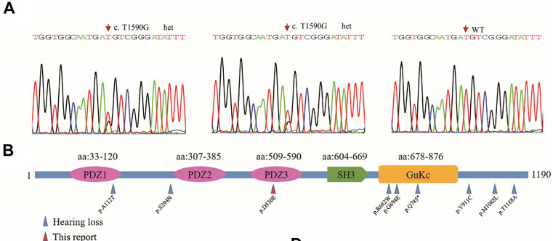
图3 TJP2基因突变分析。(A) Sanger测序色谱图显示受影响的儿子和母亲中存在杂合变异。(B) 在HGMD中治疗的所有TJP2突变均定位于编码的TJP2蛋白;蓝色箭头:在听力障碍病例中检测到的变异;红色箭头:本研究中检测到PDZ3结构域中的p.D530E。另一个导致听力障碍的变异(p.A112T)被映射到PDZ1结构域

在家庭2受影响的配偶中发现的KIF11基因新变异
家庭2中受影响的配偶经WES分析确定了一种KIF11基因的错义变异(c.733A>G,p.M245V)。据预测,这种变异是致病的,未记录在gnomAD或ChES数据库中。在小头畸形、伴有或不伴有脉络膜视网膜病变、淋巴水肿或精神发育迟滞的患者中,已经报告了至少100种不同的突变(MIM:152950和HGMD)。我们发现,KIF11中两个已报道的听力障碍相关变异(c.704C>G,pS235C和c.730C>T,pH244Y)9、10和本报告中发现的变异(c.733A>G,p.M245V)(图4A)都聚集在驱动蛋白马达结构域中的一个小区域(图4B)。推测该区域可能参与包括耳蜗细胞在内的中枢神经系统组织中的蛋白质相互作用。根据ACMG分类,该变异为PM2和PP3。因此,被归类为意义未明的变异。
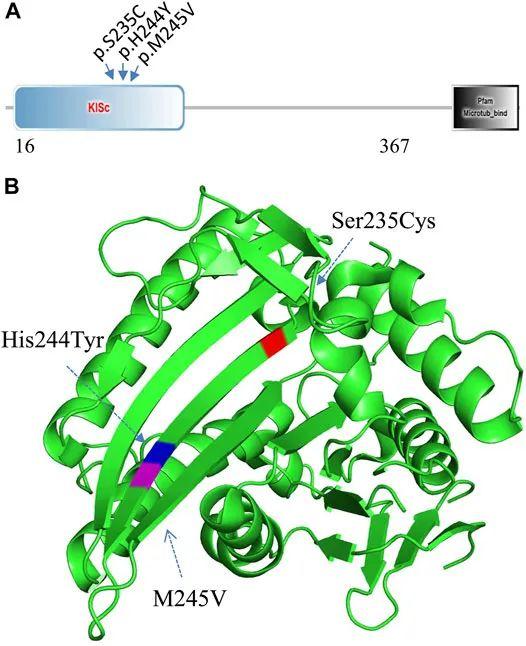
图4 KIF11基因突变分析。(A) 三种听力障碍相关变异位于KISc域(SmartMotif)。(B) 基于人类KIF11驱动蛋白结构域3D结构,三种听力障碍相关变异均位于驱动蛋白马达结构域个单个β-折叠片的一个小区域

参考文献1.Morton, C. C., and Nance, W. E. (2006). Newborn Hearing Screening - A Silent Revolution. N. Engl. J. Med. 354, 2151–2164.2.Tseng, C., and Lalwani, A. (2000). Cracking the Auditory Genetic Code: Part II. Syndromic Hereditary Hearing Impairment. Am. J. Otolaryngol. 21, 437–451.3.Gettelfinger, J. D., and Dahl, J. P. (2018). Syndromic Hearing Loss: A Brief Review of Common Presentations and Genetics. J. Pediatr. Genet. 7, 1–8.4.Yuan, Y., Zhang, J., Chang, Q., Zeng, J., Xin, F., Wang, J., et al. (2014). De Novo mutation in ATP6V1B2 Impairs Lysosome Acidification and Causes Dominant Deafness-Onychodystrophy Syndrome. Cell Res 24, 1370–1373.5.Zádori, D., Szalárdy, L., Reisz, Z., Kovacs, G. G., Maszlag-Török, R., Ajeawung, N. F., et al. (2020). Clinicopathological Relationships in an Aged Case of DOORS Syndrome with a p.Arg506X Mutation in the ATP6V1B2 Gene. Front. Neurol. 11, 767.6.Feinmesser, M., and Zelig, S. (1961). Congenital Deafness Associated with Onychodystrophy. Arch. Otolaryngol. - Head Neck Surg. 74, 507–508.7.Kim, M.-A., Kim, Y.-R., Sagong, B., Cho, H.-J., Bae, J. W., Kim, J., et al. (2014). Genetic Analysis of Genes Related to Tight junction Function in the Korean Population with Non-syndromic Hearing Loss. PLoS One 9, e95646.8.Walsh, T., Pierce, S. B., Lenz, D. R., Brownstein, Z., Dagan-Rosenfeld, O., Shahin, H., et al. (2010). Genomic Duplication and Overexpression of TJP2/ZO-2 Leads to Altered Expression of Apoptosis Genes in Progressive Nonsyndromic Hearing Loss DFNA51. Am. J. Hum. Genet. 87, 101–109.9.Jones, G. E., Ostergaard, P., Moore, A. T., Connell, F. C., Williams, D., Quarrell, O., et al. (2014). Microcephaly with or without Chorioretinopathy, Lymphoedema, or Mental Retardation (MCLMR): Review of Phenotype Associated with KIF11 Mutations. Eur. J. Hum. Genet. 22, 881–887.10.Mirzaa, G. M., Enyedi, L., Parsons, G., Collins, S., Medne, L., Adams, C., et al. (2014). Congenital Microcephaly and Chorioretinopathy Due to de novo heterozygousKIF11mutations: Five Novel Mutations and Review of the Literature. Am. J. Med. Genet. 164, 2879–2886.

北京安琪尔基因医学科技有限公司--基因检测;全外显子测序;基因医学分析
Gene4HL |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-5-27 21:10
发表于 2025-5-27 21:10


 提升卡
提升卡


